Help:
RNAlien is a tool for automatic construction of RNA family models from a single sequence. The sections of this help document cover
the use of the webservice and the commandline tool, as well as details
of the pipeline used in the backend.
If you look for source code or a installation guide for the tool please refer to the Tool subpage.
Please note that constructions can take up to 24h, if you want to
construct multiple families please use the tool instead of the webservice.
This manual is included with the Tool as manual.pdf.
Table of contents:
Webservice:
Input:
AAUUGAAUAGAAGCGCCAGAACUGAUUGGGACGAAAAUGCUUGAAGGUGAAAUCCCUGAA
AAGUAUCGAUCAGUUGACGAGGAGGAGAUUAAUCGAAGUUUCGGCGGGAGUCUCCCGGCU
GUGCAUGCAGUCGUUAAGUCUUACUUACAAAUCAUUUGGGUGACCAAGUGGACAGAGUAG
UAAUGAAACAUGCUU
Output:

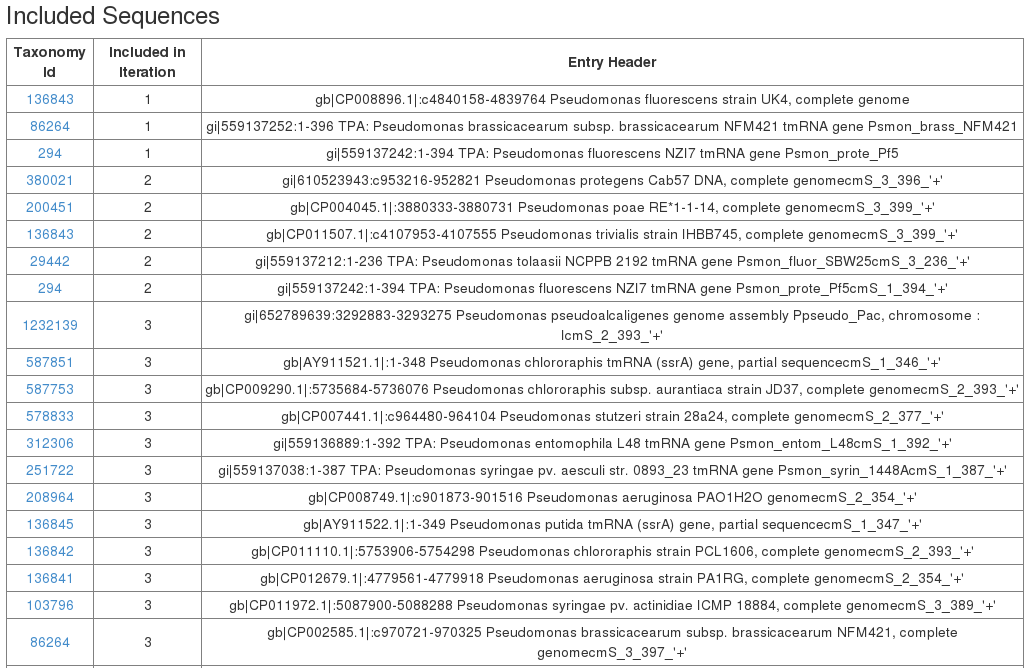
Once the construction is finished all result files are available in the result table. The output contains only the sequences (and corresponding organisms) that actually were used to construct the final RNA family model. If you want to find all sequences covered by the result model scan your database of choice with cmsearch.
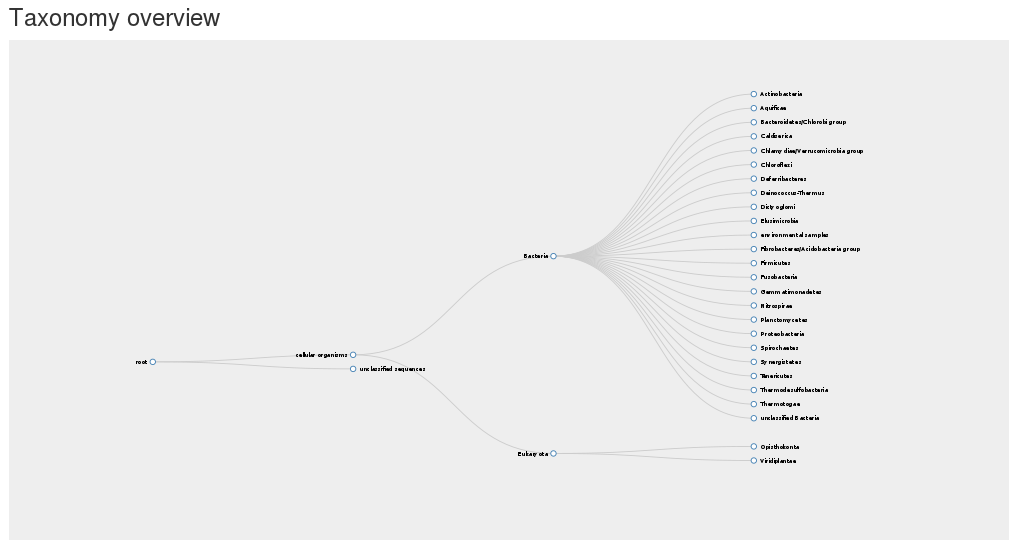
The organisms of found non-redundant sequences used in the model are visualized in the taxonomic overview.

The evaluation results for the constructed covariance model and the stockholm alignment used to build it are summarized in the evaluation table.
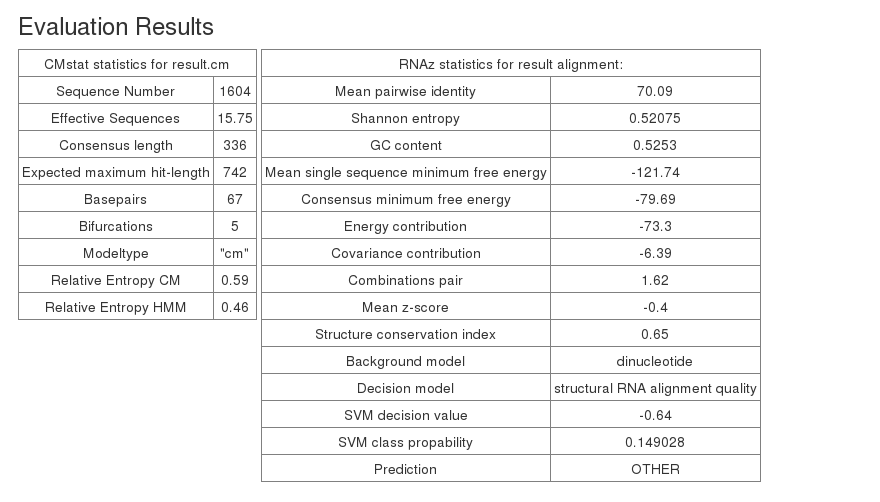
The taxonomic tree of the organsims the included sequences originated from can be found in the zoom and collapsable taxonomy tree
Finally all included sequences are listed in the included sequences table.
Tool:
Input:
| Parameter | Switch | Required | Description | Default value |
|---|---|---|---|---|
| inputFastaFilePath | -i | yes | Path to input fasta file | |
| outputPath | -o | yes | Path to output directory | |
| inputTaxId | -t | no | NCBI taxonomy ID number of input RNA organism | |
| inputnSCICutoff | -z | no | Only candidate sequences with a nSCI higher than this value are accepted. | 1.0 |
| inputEvalueCutoff | -e | no | Evalue cutoff for cmsearch filtering. | 0.001 |
| inputBlastDatabase | -b | no | Specify name of blast database to use. | nt |
| coverageFilter | -a | no | Filter blast hits by coverage of at least 80%. | True |
| singleHitperTax | -s | no | Only the best blast hit per taxonomic entry is considered. | False |
| blastsoftmasking | -f | no | Toggles blast softmasking, meaning exclusion of low complexity (repetative) regions in lookup table. | True |
| inputqueryselectionmethod | -m | no | Method for selection of queries (filtering,clustering). | filtering |
| threads | -c | no | Number of available cpu slots/cores. | 1 |
| taxonomyRestriction | -r | no | Restrict search space to taxonomic kingdom (bacteria,archea,eukaryia). | |
| sessionIdentificator | -d | no | Optional session id that is used instead of automatically generated one. | |
| Help | -? | no | Print this help |
Example call for RNAlien:
This command starts RNAlien with the input fasta file /home/user/newrna.fa, 5 cores, the taxonomy id set to E. coli. RNAlien will create a directory called construction1 in /home/user/temp/. On how to obtain the taxonomy id for the organism the sequence originates from see Retrieve Taxonomy id.
Output:
The output contains only the non-redundant sequences (and corresponding organisms) that were actually were used to construct the final RNA family model. If you want to find all sequences covered by the result model scan your database of choice with cmsearch.
+RNAlien output folder
|
|--Log file: Summary for construction process, tool versions, iteration info, evaluation results (see Log file)
|--result.cm: Result covariance model
|--result.stockholm: Result stockholm alignment
|--result.fa: Result fasta
|--result.csv
|--evaluation: contains RNAz and cmstat output for result-files
|--log: contains non-iteration specific log files
|--1 iteration directories (see description below)
|--2 ..
|--3 ..
+iterationdirectory: initial model construction
|
|--model.cm: Result covariance model
|--model.stockholm: Result stockholm alignment
|--model.fa: Result fasta
|--log: Raw and processed blast hits and accepted /rejected candidates (see iteration log directory)
|--input.fa: The input fasta sequence |--input.fold: RNAfold output of the input fasta sequence |--1.fa: fasta file for first candidate |--1.alifold: RNAalifold file for the input sequence and the first candidate
|--1.fold: RNAfold output for the first candidate
|--2.fa: Fasta file for the second candidate
|-..
+iterationdirectory: modelexpansion
|
|--model.cm: Result covariance model
|--model.stockholm: Result stockholm alignment
|--model.fa: Result fasta
|--log: Directory that contains raw and processed blast hits (see blastdirectory)
|--1.fa: fasta file for first candidate
|--1.cmsearch
|--2.fa
|--2.cmsearch
|--..
+iteration log directory
|
|--1_1blastOutput: Raw blast output for first query, indicated by leading 1_
|--1_2blastHits: Parsed blast output
|--1_3blastHitsFilteredByLength: Blasthits filtered by exceeding 3* query length
|--1_3ablastHitsFilteredByLength: Blasthits filtered by having >80% coverage
|--1_4blastHitsFilteredByParentTaxId: Only one blasthit per parent taxid
|--1_5filteredBlastResult: Only one blasthit per taxid
|--1_6requestedSequenceElements: Blasthit derived sequences requested from Entrez
|--1_10afullSequencesWithSimilars: Sequences retrieved from Entrez
|--1_10fullSequences: fullSequencesWithSimilars filtered for only containing unique sequences
|--2_1blastOutput: Raw blast output for second query, indicated by leading 2_
|--..
|--11candidates: All query specific sequences merged
|--12candidatesFilteredByCollected: Filter for sequences not identical with collected
|--13selectedCandidates: Sequences selected either by nSCI or cmsearch for inclusion in model by set evalue cutoff
|--14rejectedCandidates: Sequences that were rejected
|--15potentialCandidates: Sequences that are within a 10^3 interval of the set evalue cutoff,
will be reevaluated at end of modelconstruction
Log file
Benchmark
RNAfamilies constructed for benchmark are available as archive:
| Rfam id | Family name | Archive link (.tar.gz) |
|---|---|---|
| RF00001 | 5S rRNA | archive |
| RF00002 | 5_8S rRNA | archive |
| RF00003 | U1 | archive |
| RF00004 | U2 | archive |
| RF00005 | tRNA | archive |
| RF00008 | Hammerhead_3 | archive |
| RF00010 | RNaseP_bact_a | archive |
| RF00011 | RNaseP_bact_b | archive |
| RF00017 | Metazoa_SRP | archive |
| RF00023 | tmRNA | archive |
| RF00026 | U6 | archive |
| RF00028 | Intron_gpI | archive |
| RF00029 | Intron_gpII | archive |
| RF00032 | Histone3 | archive |
| RF00037 | IRE_I | archive |
| RF00044 | Phage_pRNA | archive |
| RF00050 | FMN | archive |
| RF00059 | TPP | archive |
| RF00114 | S15 | archive |
| RF00162 | SAM | archive |
| RF00164 | s2m | archive |
| RF00167 | Purine | archive |
| RF00168 | Lysine | archive |
| RF00169 | Bacteria_small_SRP | archive |
| RF00174 | Cobalamin | archive |
| RF00175 | HIV-1_DIS | archive |
| RF00177 | SSU_rRNA_bacteria | archive |
| RF00207 | K10_TLS | archive |
| RF00209 | IRES_Pesti | archive |
| RF00234 | glmS | archive |
| RF00374 | Gammaretro_CES | archive |
| RF00380 | ykoK | archive |
| RF00458 | IRES_Cripavirus | archive |
| RF00480 | HIV_FE | archive |
| RF00500 | TCV_H5 | archive |
| RF00504 | Glycine | archive |
| RF00843 | mir-228 | archive |
| RF00871 | mir-689 | archive |
| RF01051 | c-di-GMP-I | archive |
| RF01054 | preQ1-II | archive |
| RF01073 | GP_knot1 | archive |
| RF01118 | PK-G12rRNA | archive |
| RF01380 | HIV-1_SD | archive |
| RF01510 | MFR | archive |
| RF01689 | AdoCbl-variant | archive |
| RF01734 | crcB | archive |
| RF01786 | c-di-GMP-II | archive |
| RF01831 | THF | archive |
| RF01852 | tRNA-Sec | archive |
| RF01856 | Protozoa_SRP | archive |
| RF01857 | Archaea_SRP | archive |
| RF01998 | group-II-D1D4-1 | archive |
| RF02001 | group-II-D1D4-3 | archive |
| RF02095 | mir-2985-2 | archive |
| RF02253 | IRE_II | archive |
| RF02519 | ToxI | archive |
Values for the specificity and recall plots were computed with the RNAlienStatistics executable and the rnalienstatistics.pl script included in RNAlien.
RNAalien pipeline
RNAlien pipeline
Retrieve Taxnonomy id
Enter the organisms name in the Taxonomy field (e.g Escherichia coli) and click search. You are then redirected to a Summary page. Click the organism name. Depending on how specific your search was you can either select from a list of organisms or you are directed to a organism page Escherichia coli str. K-12 substr. MG1655star. You can find the taxonomy id right below the organisms name (Escherichia coli str. K-12 substr. MG1655star Taxonomy ID: 879462).